或者有的gff最后部分是scaffold部分,需要删掉吗
基因组基因复制的分类鉴定:DupGen-finder参考:https://www.jianshu.com/p/0a14d971bd0a[https://www.jianshu.com/p/0a14d971bd0a] requi...
或者有的gff最后部分是scaffold部分,需要删掉吗
基因组基因复制的分类鉴定:DupGen-finder参考:https://www.jianshu.com/p/0a14d971bd0a[https://www.jianshu.com/p/0a14d971bd0a] requi...
或者后面会有scafford部分,需要删掉嘛
「基因组」基因组分析之鉴定基因类型基因类型鉴定 基因组的组装和注释完成,得到配套的基因组和注释文件之后,可以利用gffread软件提取对应的gene的cds,pep序列。应运而生,下游的生信或者实验想知道关注...
我想问下博主大大gff文件有的在后面会有未鉴定出染色体组的那部分,需要删掉这些吗
「基因组」基因组分析之鉴定基因类型基因类型鉴定 基因组的组装和注释完成,得到配套的基因组和注释文件之后,可以利用gffread软件提取对应的gene的cds,pep序列。应运而生,下游的生信或者实验想知道关注...
我想问下gff文件有的在后面会有未鉴定出染色体组的那部分,需要删掉这些吗
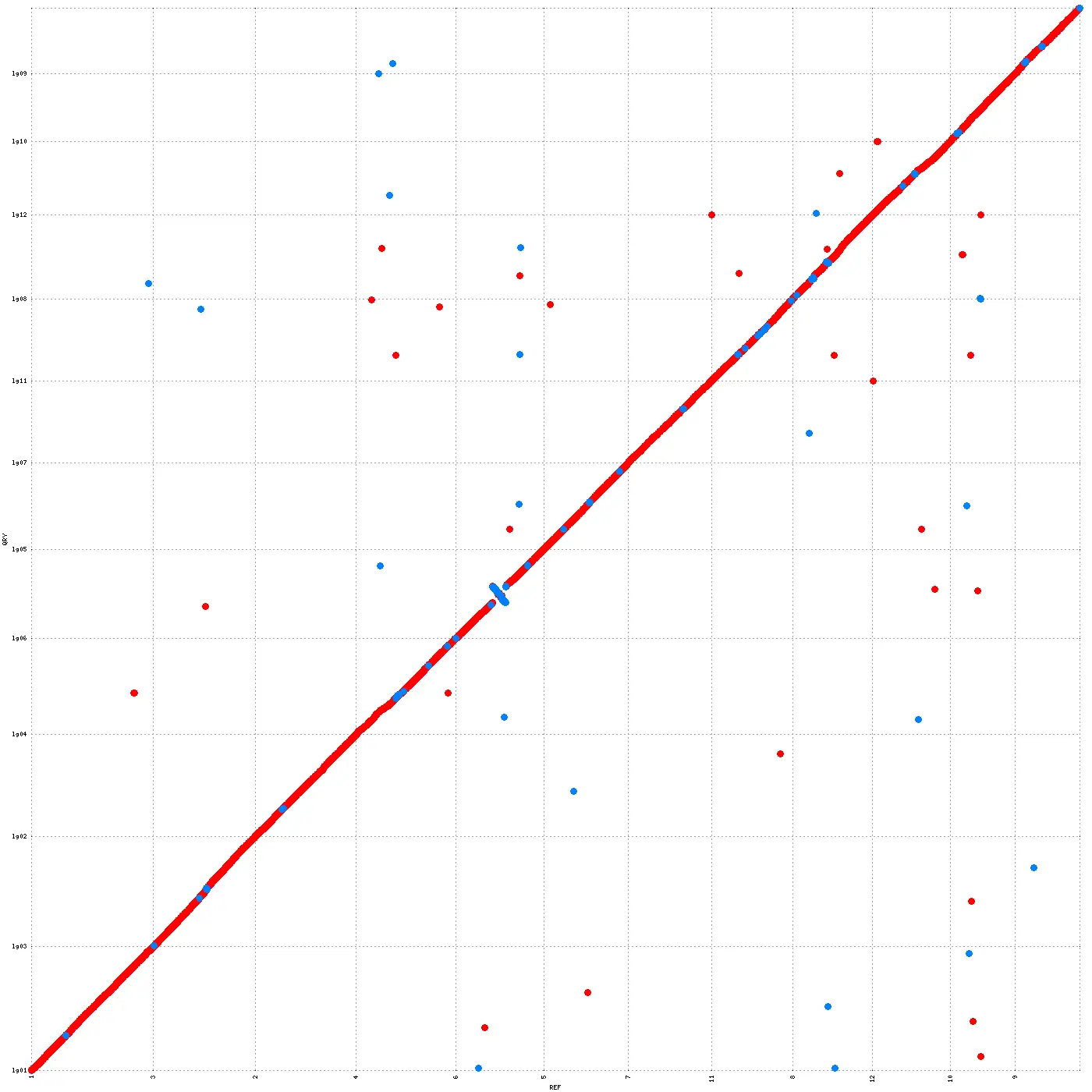
DupGen_finder安装及使用串联复制(Tandem duplication) :串联复制主要发生在染色体重组区域,串联复制形成的基因家族成员通常紧密排列在同一条染色体上,形成一个序列相似、功能相近的基因...
我想问下gff文件有的在后面会有未鉴定出染色体组的基因,需要删掉这些吗
基因组基因复制的分类鉴定:DupGen-finder参考:https://www.jianshu.com/p/0a14d971bd0a[https://www.jianshu.com/p/0a14d971bd0a] requi...
@沉_0cb2 你好,我是把七百多条序列的fasta文件放入查找,设置的是5条motif,结果也出来了,但是各个motif的功能什么的信息都没有呀,那怎么分析
MEME在线网站查找motif步骤小白日记第一天,记录一下自己查找motif的步骤,以便日后忘记了再重学。之前也用过许多软件,都是在要用的时候上网上各种搜索操作步骤,边学边用,只顾获得结果了,从未意识到要将操...
@BeeBee生信 不是研究人的话是不是可以不做这一步,直接用转录组数据跑,然后libtype用A可以嘛
Salmon 进行转录本定量Salmon 是转录本定量软件,使用转录本定量主要优点一是更准确,比如不同样本同一基因使用不同 isoform 此时基因长度并不相等,直接基因定量无法顾及;二是方便进行可变剪...
你好,如果基因组没有标明primary怎么办,就像ncbi里直接就写的genomic.fna
快速计算基因表达软件:Salmon我们常见的转录组表达分析一般都是将reads比对至参考基因组或者转录组上,然后在基因或者转录本水平上定量表达丰度。 但最近在做小RNA分析时却遇到了没有参考基因组注释文件(g...

到底还是写了一篇推文,却着实想不到一个合适的题目。可能这是我写过最长的一篇推文,约莫万字。这篇推文的主要内容应该包括: TBtools 开发为了什么? TBtools 是怎么...

写在前面 做生信数据分析时,最常遇到的问题,仍然是文本处理。主要原因简单,我们永远不知道上一个人会给我们什么样的东西,而我们要的又常常不是他们给的。基于 GFF 提取 CDS...
@Charon_7db5 可以的,利用基因组fa和gff文件提取
生信小白如何在半年收到核心期刊录用证明顺利毕业!!!(基因家族成员的确定)一篇生信家族的文章比较重要的一步是家族成员的确定,所有基本分析和进化分析都是基于此。 前面推文提到本人的参考物种是模式植物拟南芥,因此我首先下载了很多关于拟南芥的综述类和研究...
请问博主可以直接用gffread提取的pep序列吗
生信小白如何在半年收到核心期刊录用证明顺利毕业!!!(基因家族成员的确定)一篇生信家族的文章比较重要的一步是家族成员的确定,所有基本分析和进化分析都是基于此。 前面推文提到本人的参考物种是模式植物拟南芥,因此我首先下载了很多关于拟南芥的综述类和研究...

关于 TBtools,我们已经聊得太多。前前后后应该也有数百篇推文,正好对应了 TBtools 数百个功能。我也曾经提过,推文当然好,但能起的作用,是宣传。如果说有记录的功能...

1.使用官方的工具包 SRA Toolkit 下载 01. Downloading SRA Toolkit · ncbi/sra-tools Wiki (github.com...
下载NCBI数据,使用ascp命令是最快的,但是小编之前尝试用该命令下载SRA数据,查阅了百度上的各种参数,最后还是没有成功,可能是有个“墙”存在的原因,小编后来有一段时间使...